ГЛАВА 333. НАРУШЕНИЯ ПОЛОВОЙ ДИФФЕРЕНЦИРОВКИ
Джин Д. Вилсон, Джеймс Е. Гриффин Ш (JeanD. Wilson, JamesE. GriffinШ)
Половая дифференцировка — последовательный и упорядоченный процесс. Хромосомный пол, формирующийся в момент оплодотворения, определяет гонадиый пол, а гонадный пол в свою очередь обусловливает развитие фенотипического пола, предполагающего образование мужского или женского мочеполового аппарата (табл.333-1). Изменения на любом этапе этого процесса во время эмбриогенеза приводят к нарушениям половой дифференцировки. К знаменитым причинам нарушения полового развития относятся изменения окружающей среды, например, при приеме вирилизирующих средств во время беременности, несемейные аберрации половых хромосом, например 45, Х-дисгенезия гонад, врожденные пороки развития многофакторного генеза. например большинство случаев гипоспадии, а также потомственные дефекты, обусловленные мутациями одиночных генов, например синдром тестикулярной феминизации.
Ограниченность знаний дозволяет дать лишь эмпирическую оценку характеру физиологических нарушений при некоторых недостатках. Тем не менее с поддержкою комбинации методов генетического, (фенотипического н хромосомного анализа обычно удается установить конкретный диагноз, определить пол и в нужных случаях произвести изменения фенотипа.
Нормальная половая дифференцировка
На первом этапе половой дифференцировки устанавливается хромосомный пол: пол гетерогаметы (XY)— мужской, а гомогаметы (XX) — женский. Затем примерно до 40-го дня беременности эмбрионы обоего пола развиваются одинаково. Вторая стадия половой дифференцировки содержится в превращении недифференцированных гонад в яички или яичники. Дифференцировка гонад в яички опосредуется генами Y-хромосомы, один из которых либо идентичен гену, кодирующему HY-антиген, либо узко сцеплен с ним. Завершающий процесс — трансляция гонадного пола в фенотипический пол — зависит от типа образовавшихся гонад плода и их эндокринной секреции. Развитие фенотипического пола приводит к (формированию мужского и женского мочеполового аппарата.
Внутренние половые органы образуются из вольфовых и мюллеровых протоков, которые на ранних стадиях эмбрионального развития обоих полов расположены рядом (рис. 333-1, а). У зародышей мужского пола вольфовы протоки дают начало придаткам яичка, ссмявыносящим протокам и семенным пузырькам, а мюллеровы протоки пропадают. У эмбрионов женского пола из мюллеровых протоков развиваются маточные трубы, матка и верхняя часть влагалища, а вольфовы протоки регрессируют. Внешние гениталии и уретра у плодов обоего пола развиваются из общей закладки — урогенитального синуса и полового бугорка, складок и вздутий (рис.333-1, б). Урогенитальный синус у плода мужского пола дает начало предстательной железе и простатической доли уретры, а у плода женского пола —уретре и доли влагалища. Из полового бугорка образуется головка полового члена у плодов мужского пола и клитор у плодов женского пола. Урогенитальные вздутия превращаются в мошонку или великие половые губы, а половые складки — в малые половые губы или сливаются, образуя мужскую уретру и ствол полового члена.
Если яички отсутствуют, как, например, у нормальных эмбрионов женского пола или у мужских зародышей, кастрированных до начала фенотипической дифференцировки, развитие фенотипического пола происходит по женскому направлению. Таким образом. маскулинизация плода —это положительный результат деянья гормонов эмбриональных половых желез, тогда как развитие по женскому типу не требует пребыванья гонад. Половой фенотип в норме подходит хромосомному полу. Иными словами, хромосомный пол определяет гонадный пол, а гонадный пол в свою очередь контролирует фенотипический пол.
Таблица 333-1.Классификация нарушений полового развития у человека
Нарушения хромосомного пола Синдром Клайнфелтера Мужчины с кариотипом XX Дисгенезия гонад Смешанная дисгенезия гонад Правильный гермафродитизм Нарушения гонадного пола Чистая дисгенезия гонад Синдром неименья тестикул Нарушения фенотипического пола Женский псевдогермафродитизм
Врожденная гиперплазия надпочечников Женский псевдогермафродитизм вненадпочечникового генеза Нарушения развития мюллеровых протоков Мужской псевдогермафродитизм Нарушения синтеза андрогенов Нарушения деянья андрогенов Синдром персистенции мюллеровых протоков Нарушение развития мужских гениталий
Формирование мужского фенотипа обусловливается деянием трех гормонов. Два из них — вещество, ингибирующее мюллеровы протоки, и тестостерон — это секреторные продукты эмбриональных яичек. Вещество, ингибирующее мюллеровы протоки, представляет собой белковый гормон, который вызывает обратное развитие мюллеровых протоков и, следовательно, предотвращает формирование матки и маточных труб у эмбрионов мужского пола. Тестостерон непосредственно стимулирует дифференцировку производных вольфовых протоков и служит предшественником третьего эмбрионального гормона — дигидротестостерона (см. гл.330). Дигидротестостерон, который образуется из присутствующего в крови тестостерона, индуцирует формирование мужской уретры, предстательной железы, полового члена и мошонки. Таким образом, во время внутриутробной жизни тестостерон и дигидротестостерон вызывают образование акцессорных органов мужской репродуктивный системы, действуя через тот же внутриклеточный механизм, который опосредует их эффекты в дифференцированных тканях (см. гл.330).
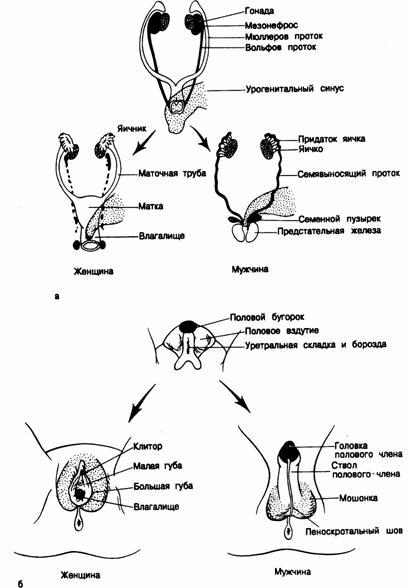
Рис.333-1. Нормальная половая дифференцировка. а — внутренние половые органы; б — внешние половые органы.
Секреция тестостерона эмбриональными тестикулами достигает максимума к 8— 10-й неделе беременности, а формирование полового фенотипа завершается в главном к концу I триместра. На поздних стадиях беременности у плодов женского пола происходят развитие фолликулов в яичниках и созревание влагалища, а у плодов мужского пола — опущение яичек и рост внешних гениталий.
Таблица 333-2.Клинические проявления нарушений хромосомного пола
|
Нарушение |
Хромосомные аберрации |
Развитие гонад |
Наружные гениталии |
Внутренние гениталии |
Развитие молочных желез |
Примечания |
|
Синдром Клайнфелтера |
47,ХХY или 46, XY/ 47,XXY |
Гиалинизированные яички |
Нормальные мужские |
Нормальные мужские |
Гинекомастия |
Самое частое нарушение половой дифференцировки, высокий рост |
|
Мужчина с кариотипом XX |
46, XX |
То же |
То же |
То же |
То же |
Рост ниже нормы для мужчин; повышенная частота гипоспадии, сходство с синдромом Клайнфелтера. Может быть семейным |
|
Дисгенезия гонад (синдром Тернера) |
45,X или 46, XX/ 45, X |
Гонадальные тяжи |
Незрелые женские |
Гипоплазированные женские |
Незрелые женские |
Низкорослость и множественные соматические аномалии. Может быть 46,XX со структурными нарушениями Х-хромо-сомы |
|
Смешанная дисгенезия гонад |
46, XY/ 45,X или 46, XY |
Яички и гонадальные тяжи |
Варьируют, но почти всегда сомнительны;60% воспитываются как девочки |
Матка, влагалище и одна маточная труба |
Обычно по мужскому типу |
Вторая по распространенности причина сомнительных гениталий у новорожденных; часты опухоли Может быть семейным |
|
Истинный гермафродитизм |
46, XX или 4б,ХYили мозаицизм |
Яички и яичники или овотестис |
Варьируют, но обычно сомнительны; 60% воспитываются как мальчики |
Обычно матка и урогенитальный синус; протоки подходят гонадам |
Гинекомастия в 75% случаев |
|
Нарушения хромосомного пола
Нарушения хромосомного пола (табл.333-2) возникают при изменении числа или строения Х- или Y-хромосом (см. гл.60).
Синдром Клайнфелтера
Клинические проявления.Синдром Клайнфелтера характеризуется первичным гипогонадизмом (махонькие твердые яички), азооспермией, гинекомастией и повышенным уровнем гонадотропинов в плазме мужчин с двумя или более Х-хромосомами. Кариотип — чаще 47, XXY (классическая форма) или мозаицизм 46, XY/47, XXY. Этот синдром представляет собой самое распространенное нарушение половой дифференцировки и встречается с частотой примерно 1 случай на 500 мужчин.
В препубертатном возрасте у больных отмечают махонькие тестикулы, но в остальном они выглядят нормально. После полового созревания хворь проявляется бесплодием, гинекомастией или иногда недостаточной андрогенизацией (табл.333-3). Постоянным признаком кариотипа 47, XXY является гиалинизация семенных канальцев и азооспермия. Яички махонькие, плотные, длиной менее
По результатам исследования хромосомного кариотипа в лейкоцитах периферической крови установлено, что мозаичным вариантом синдрома страдают около 10% больных. Частота этого варианта, по-видимому, занижена, так как хромосомный мозаицизм может иметь место только в тестикулах, а кариотип периферических лейкоцитов — оставаться нормальным. Мозаичная 4юрма протекает обычно не столь тяжело, как вариант 47, XXY, и яички могут хранить нормальные размеры (см. табл.333-3). Эндокринные нарушения также выражены слабее, а гинекомастия и азооспермия встречаются реже. Больше того, больные с мозаицизмом иногда могут хранить фертильность. У некоторых из них из-за незначительности физических отклонений от нормы можно и не подозревать правильного диагноза.
Таблица 333-3.Характеристика больных с классическим и мозаичным вариантом синдрома Клайнфелтер1
|
Признак |
47, XXY, % |
46, XY/47, X XY,% |
|
Изменение гистологии яичек |
100 |
942 |
|
Уменьшение длины яичек |
99 |
732 |
|
Азооспермия |
93 |
502 |
|
Сниженный уровень тестостерона |
79 |
33 |
|
Уменьшение роста волос на лице |
77 |
64 |
|
Повышенный уровень гонадотропинов |
75 |
332 |
|
Сниженная половая функция |
68 |
56 |
|
Гинекомастия |
55 |
332 |
|
Уменьшение роста волос под мышками |
49 |
46 |
|
Уменьшенная длина полового члена |
41 |
21 |
1
Таблица основана на результатах обследования 519 больных с кариотипом XX и 51 с кариотипом XY/XXY— больного. 2 Вероятность различий р <0,05 или еще выше. Из Gordon et al.

Рис.333-2. Схема нормального сперматогенеза и оплодотворения.
Показаны эффекты нерасхождения в мейозе и митозе, что приводит к формированию классического синдрома Клайнфелтера, синдрома Тернера и мозаичной формы синдрома Клайнфелтера. Схема не изменится, если нарушения будут происходить в процессе оогенеза.
Описано еще около 30 вариантов кариотипа при синдроме Клайнфелтера, как без мозаицизма (XXYY, XXXYи XXXXY), так и с мозаицизмом с сопутствующими структурными нарушениями Х-хромосомы или без них. Как правило, чем больше степень хромосомных нарушений (а при мозаичной форме — чем больше патологических клеточных линий), тем более тяжелы клинические проявления.
Патофизиология.Классическая форма обусловливается нерасхождением хромосом в мейозе в процессе гематогенеза (рис.333-2). Примерно в 40% случаев нерасхождение в мейозе происходит при сперматогенезе, а в 60%— при оогенезе. С увеличением возраста матери вероятность нерасхождения увеличивается. Мозаичную форму относят за счет нерасхождения хромосом в митозе после оплодотворения яйцеклетки; это нерасхождение может иметь место как в 46, XY-зиготе, так и в 47, XXY-зиготе. Двойной дефект (нерасхождение и в мейозе и в митозе) чаще всего служит причиной синдрома и объясняет тем самым, почему его мозаичная форма диагностируется реже, чем классическая.
Содержание фолликулостимулирующего (ФСГ) и лютеинизирующего гормона (ЛГ) в плазме обычно повышено; из-за постоянного дефекта семенных канальцев уровень ФСГ меньше перекрывается нормальными показателями и имеет большее диагностическое значение. Уровень тестостерона в плазме в среднем составляет половину нормального, но его колебания перекрываются нормальными. Среднее содержание эстрадиола в плазме по не совсем ясным причинам повышено. На ранних этапах заболевания яички могли бы секретировать большие количества эстрадиола вследствие повышенного уровня ЛГ в плазме, но в конце концов тестикулярная секреция эстрадиола (и тестостерона) снижается. Повышение содержания эстрадиола на поздних стадиях заболевания можно объяснить, вероятно, сочетанием уменьшения скорости его метаболического клиренса с ускорением конверсии тестостерона в эстрадиол вне железы. В результате как на ранних, так и на поздних стадиях проявляется та или иная степень недостаточной андрогенизации и избыточной феминизации. Феминизация, включая гинекомастию, зависит от относительного или абсолютного преобладания эстрогенов над андрогенами в крови, и у лиц с меньшим содержанием тестостерона и большим содержанием эстрадиола возрастает вероятность развития гинекомастии (см. гл.332). Повышение содержания гонадотропинов в плазме после введения рилизинг-гормона лютеинизирующего гормона (ЛГРГ) в постпубертатном возрасте более значительное, а нормальное ингибирующее действие тестостерона на гипофизарную секрецию ЛГ (отрицательная обратная связь) ослаблено. У больных с нелеченым синдромом Клайнфелтера может иметь место «реактивная патология гипофиза» в виде увеличения или деформации турецкого седла. Это объясняется, по-видимому, хроническим выпадением влияния гонад по механизму отрицательной обратной связи и гипертрофией гонадотрофов вследствие их стимуляции ЛГРГ. Возникает ли в таких случаях настоящая аденома, неизвестно.
Лечение.Восстановить фертильность при синдроме Клайнфелтера невозможно, а единственным эффективным способом коррекции гинекомастии является хирургическое удаление ткани молочных желез. Некоторым больным с недостаточной андрогенизцией помогает терапия андрогенами, но иногда она приводит к парадоксальному усилению гинекомастии, вероятно, за счет того, что увеличивает доступность субстратов для образования эстрогенов в периферических тканях. Андрогены следует применять в форме тестостерона ципионата или тестостерона энантата. При введении тестостерона уровень ЛГ в плазме, если и нормализуется, то лишь через несколько месяцев.
Синдром ХХ-мужчины
Кариотип 46, XX у фенотипических мужчин встречается с частотой приблизительно 1:20000 — 1:24000. У таких лиц все женские внутренние гениталии отсутствуют, и в психосексуальном плане они ощущают себя мужчинами. Действительно, признаки этого синдрома сходны с таковыми при синдроме Клайнфелтера: яички маленькие и плотные (обычно меньше
Патогенез этого нарушения объясняют следующим образом:1) транслокацией части Y-хромосомы на Х-хромосому;2) мозаицизмом по Y-хромосоме в некоторых клеточных линиях или ранней потерей Y-хромосомы; 3) мутацией аутосомного гена и 4) делецией генетического вещества Х-хромосомы, в норме оказывающего отрицательный регуляторный эффект на развитие яичек. Однако ни одна из них не в состоянии полностью объяснить данное нарушение. Мозаицизм в большинстве случаев вряд ли имеет место, но все остальные процессы вполне возможны. Нельзя исключить и гетерогенную природу синдрома. Терапевтические мероприятия в таких случаях аналогичны таковым при синдроме Клайнфелтера.
Дисгенезия гонад (синдром Тернера)
Клинические проявления.Дисгенезия гонад характеризуется первичной аменореей, половым инфантилизмом, низкорослостью, множественными врожденными аномалиями и наличием гонадальных тяжей с обеих сторон у фенотипических женщин с каким-либо дефектом Х-хромосомы. Это состояние следует отличать:1) от смешанной дисгенезии гонад, при которой с одной стороны имеется яичко, а с другой — гонадальный тяж;2) от чистой дисгенезии гонад: в этом случае гонадальные тяжи с обеих сторон имеют место у лиц с нормальным кариотипом 46, XX или 46, XY, нормальным ростом и первичной аменореей;3) синдрома Нунан — аутосомно-доминантного нарушения у мужчин и женщин, характеризующегося складчатой кожей на шее, низкорослостью, врожденными пороками сердца, вальгусной деформацией предплечий и другими врожденными дефектами, несмотря на нормальные кариотип и гонады.
Частота дисгенезии гонад — 1:2500 новорожденных девочек. Диагноз ставят либо сразу после рождения по сопутствующим врожденным порокам, либо, что чаще, в пубертатном возрасте, когда врожденным аномалиям сопутствует аменорея. Дисгенезия гонад — самая распространенная причина первичной аменореи (около 30%). Больные не достигают полового созревания, наружные гениталии женского типа, но недоразвитые, так же как и молочные железы (в том случае, если больная не лечилась эстрогенами). Внутренние гениталии представлены инфантильными маточными трубами и маткой; в широких связках с обеих сторон присутствуют гонадальные тяжи. В процессе эмбриогенеза транзиторно появляются примордиальные зародышевые клетки, но они исчезают в результате ускоренной атрезии (см. гл.331). К возрасту ожидаемого полового созревания эти тяжи уже не содержат различимых фолликулов и яйцеклеток; в них присутствует фиброзная ткань, неотличимая от нормальной стромы яичников.
Сопутствующие соматические аномалии затрагивают в основном скелет и соединительную ткань. В младенческом возрасте болезнь диагностируют по наличию лимфатического отека кистей и стоп, складчатости шеи, низкой линии оволосения, избыточных кожных складок на затылке, щитообразной грудной клетке с широко расставленными сосками и малой массе тела при рождении. Кроме того, у больных характерное лицо с маленькой челюстью, эпикантус, низко расположенные или деформированные уши, рыбий рог и птоз. В 50% случаев отмечают укорочение IV пястных костей, а в 10—20%—коарктацию аорты. Рост у взрослых больных редко превышает
Патофизиология.Примерно у 50% больных обнаруживают кариотип 45. X, у 25% — мозаицизм без структурных нарушений (46, ХХ/45,X). а у остальных — структурные па-рушения Х-хромосомы с мозаицизмом или без него (см. гл.60). Вариант 45, Х обусловлен потерей хромосомы в процессе гаметогенсза у любого из родителей или с ошибкой митоза при одном из ранних делений оплодотворенной зиготы (см. рис.332-2). Низкорослость и другие соматические изменения являются следствием потери генетического материала с короткого плеча Х-хромосомы. Гонадальные тяжи образуются при потере генетического материала либо с длинного, либо с короткого плеча Х-хромосомы. У больных с мозаицизмом или структурными нарушениями Х-хромосомы изменения фенотипа занимают по тяжести промежуточное положение между теми, которые наблюдаются при варианте 45,Х, и нормой. У некоторых больных с гипертрофией клитора, помимо Х-хромосомы, присутствует фрагмент еще какой-то хромосомы, предположительно — аномальной Y-хромосомы. В редких случаях сбалансированная Х-аутосомная транслокация может обусловить семейную передачу дисгенезии гонад (см. гл.60).
Раньше для выявления нарушений Х-хромосом исследовали половой хроматин. Половой хроматин (тельца Барра) у здоровых женщин — продукт инактивации одной из двух Х-хромосом; женщин с хромосомным набором,45,X, подобно нормальным мужчинам, относили к группе хроматинотрицательных. Однако хроматннотрицательными являются лишь около 50% больных с дисгенезией гонад (больные с кариотипом 45,Х и с наиболее выраженным мозаицизмом и структурными нарушениями). Поэтому для установления диагноза и идентификации больных с элементами Y-хромосомы и высоким риском возникновения злокачественных опухолей в гонадальных тяжах необходим анализ кариотипа.
В период ожидаемого полового созревания оволосение подмышек и лобка скудное, молочные железы неразвиты, менструаций нет. Содержание ФСГ в сыворотке, повышенное в младенчестве, в детстве снижается до нормы, а в возрасте 9—10 лет возрастает до уровня, характерного для кастратов. В это время содержание ЛГ в сыворотке также повышено, а уровень эстрадиола в плазме снижен (менее 10 пг/мл). Примерно у 2%>больных с вариантом 45.Х и у 12% больных с мозаициэмом в яичниках сохраняется достаточное число фолликулов, чтобы иногда возникали менструации. Больше того, у лип с минимальными повреждениями иногда вероятна беременность . Однако продолжительность детородного периода у таких больных невелика.
Лечение.В ожидаемое время полового созревания следует начать заместительную терапию эстрогенами, чтобы индуцировать развитие молочных желез, половых губ, влагалища, матки и маточных труб (см. гл.331). В первый год лечения эстрадиолом скорость роста тела в длину и созревания костей примерно удваивается, но конечный рост больных редко достигает ожидаемого (см. гл.331). Лечение гормоном роста не приносит успеха. У больных с вариантом 45, Х опухоли гонад встречаются редко, но у некоторых лиц с мозаицизмом по Y-хромосоме они возникают. Поэтому гонадальные тяжи следует удалять в любом случае при наличии признаков вирилизации пли при обнаружении линии клеток, содержащих Y-хромосому.
Смешанная дисгенезия гонад
Клинические проявления.Смешанная дисгенезия гонад —это состояние, при котором у фенотипических мужчин или женщин с одной стороны имеется яичко, а с иной — гонадальный тяж. У большинства больных обнаруживается мозаицизм 45, Х/46,XY, по клинические проявления выходят за рамки определяемых этой аберрацией хромосом. Частота синдрома безызвестна, но, по данным большинства клиник, это вторая по частоте (после врожденной гиперплазии надпочечников) причина амбисексуальности гениталий у новорожденных.
Примерно 60% больных считают